



完成人基因组的测序后,是时候卷起袖子着手解决另一项艰巨任务了,即解密复杂的蛋白质组。这样,蛋白质组学的时代、研究所有表达的蛋白的功能都时代也就降临了。这项任务尤其复杂,因为与基本都是稳定不变的人类基因组不同,蛋白质组在细胞之间都是有差异的,例如肝细胞和脑细胞之间、健康细胞和癌细胞之间、甚至同一个细胞的不同发育阶段之间。
为应对这一挑战,建立了人类蛋白质组项目。该项目的使命是通过绘制人体蛋白质分子图谱,全面解析人类基因的功能特征。由此,该项目已成为诠释基因生物学功能与分子机制的重要资源,并为疾病的先进诊断与治疗方法提供支撑。
什么是翻译后修饰 (PTM)?
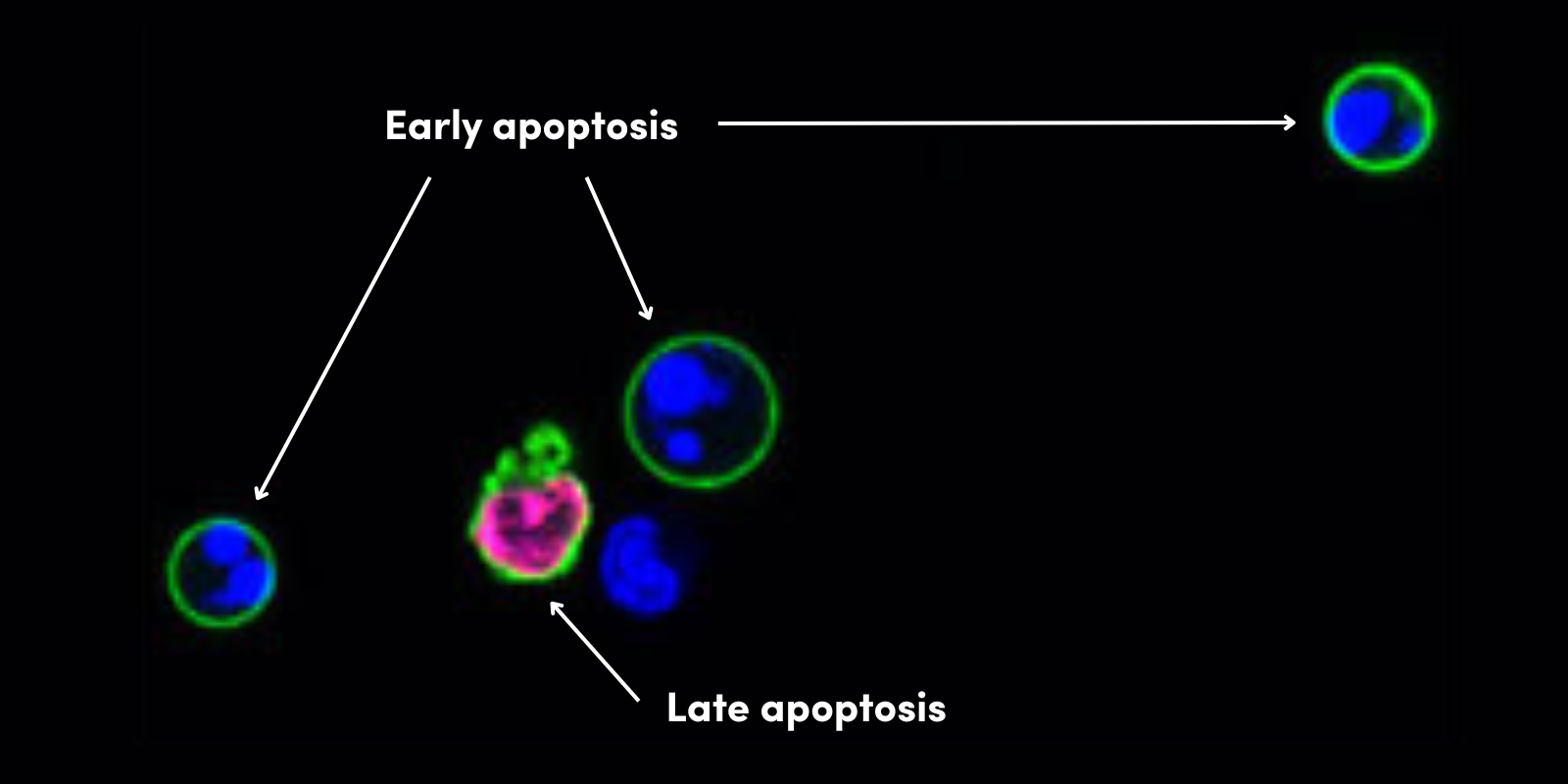
约 20,000 种检测的基因已被翻译为多达上百万种蛋白形式,或蛋白变体。那么,为什么基因数量和蛋白变体的数量之间有如此巨大的差异呢?启动子和基因选择性剪切会在一定程度上增大蛋白质组学的复杂性,但其在很大程度上是由蛋白翻译后修饰 (PTM) 所导致的。PTM 是共价修饰,包括向特定氨基酸以及溶蛋白性裂解物加入磷酰基、乙酰基、甲基、泛素基或其他基团。这些修饰是蛋白活性、亚细胞定位、降解和蛋白间相互作用所必需的。了解 PTM 在疾病状态中的作用,对检测生物标记物和开发疗法都至关重要。
自下而上与自上而下的方法
质谱法 (MS) 是一种高敏感技术,大规模地用来研究某种样品中的蛋白及其 PTM。质谱法技术的进展对蛋白质组学起到了推动作用。最成熟且应用最广的策略称为“自下而上”的蛋白质组学方法,其中,采用蛋白酶(通常为胰蛋白酶)将细胞/组织样品消化为短肽,然后再进行 MS 检测。随后,根据肽信息推断蛋白变体的特征。
“自下而上”的蛋白质组学方法是在进行质谱分析前不消化样品,这种策略的使用并不是非常广泛,但在临床研究中有潜在应用,因为可以从天然蛋白(降解产物、序列变异和 PTM 组合)的分析中获取其他信息。
MS 有很多不同的类型。用于蛋白质组学方法的一种常见类型是液相色谱串联质谱 (MS) 或 LC-MS/MS。在 LC-MS 中,消化得来的肽在加入质谱仪前用高效液相色谱法进行分离。这个仪器可以用来收集完整肽 (MS1) 及其肽片段(MS2 或 MS/MS) 极为准确的质荷比测量值。这些质荷比测量值与一个包含所有蛋白的数据库相匹配,可用来识别这种肽。
基于质谱法的蛋白质组学方法是一种强大工具,可用于检测磷酸化、泛素化和乙酰化等的新底物,检测和验证药物靶标,发现生物标记物,阐明药物脱靶效应,并探讨药物/化学调节剂的作用机制。
了解更多
在我们的蛋白质组学简介系列博客《翻译后修饰富集:基于抗体的蛋白质组学分析策略》中,我们将介绍两种自下而上的方法来识别和定量复杂生物样品中的翻译后修饰。
您可能还想观看简化蛋白质组学网络研讨会。
需要一种参考工具来显示每种氨基酸被哪种 PTM 修饰?下载我们的“氨基酸翻译后修饰”海报。




 沪公网安备31011502018823号
沪公网安备31011502018823号