


蛋白激酶在调节几乎所有细胞活动时起着至关重要的作用。通过磷酸化,这些信号转导酶就像是音乐大师,通过调节蛋白质(和其他激酶)来调整其活性、调节其稳定性、改变其亚细胞位置或标记它们以便销毁,从而协调细胞生长、分裂和代谢。
因此,应当毫不奇怪,这些重要调节性蛋白质的失调是一种常见的疾病机制,特别在癌症、神经系统疾病和发育问题等方面如此。然而,虽然我们知道人类基因组中存在超过 530 种激酶,其中超过 400 种具有催化活性,但往往难以或几乎不可能确定哪些激酶调节哪些靶标蛋白(或相应的激酶底物)。
人们早就知道,若能轻易确定导致关键底物失调的激酶,就可以揭示许多新的药物靶标并改变治疗格局,但直到现在,这完全不可能大规模实现。
现在,麻省理工学院、耶鲁医学院和威尔康奈尔医学院的研究人员已利用强大的计算工具、纯化的激酶和肽文库以及 Cell Signaling Technology 资源 (PhosphositePlus) 上可提供的数据,开发一种算法来预测负责调节特定蛋白质底物的激酶。
“激酶文库”工具现在可通过 PhoshoSitePlus 使用。
2023 年 1 月,《自然》杂志发表了一篇题为《人类丝氨酸/苏氨酸激酶组底物特异性图谱》的论文,详细介绍了该项目,重点研究了癌症中的丝氨酸/苏氨酸激酶活化状态。1 在这里,您可以了解为该项目做出贡献的开创性工作,以及与 CST 合作创建激酶库将如何帮助研究人员发现新的药物靶点。
终身追求:阐明信号转导通路中的蛋白激酶
哈佛医学院和达纳法伯研究所 Lewis “Lew” Cantley 教授在这项研究期间系威尔康奈尔医学院教授,已专注于激酶信号转导数十年。Cantley 教授以其在信号转导和新陈代谢方面的开创性发现(包括鉴定 PI3 激酶)而闻名,他的工作对我们理解癌症生物学和疾病进展已产生重大影响。
几十年来,像 Cantley 教授这样的蛋白质组学研究人员已经使用质谱 (MS) 鉴定到癌细胞中磷酸化程度高于健康细胞的蛋白质。然而,基于 MS 的磷酸化蛋白质组学仅提供有关磷酸化蛋白质的信息,并不能鉴定导致这些磷酸化事件的直接激酶。此外,询问 MS 数据以确定潜在磷酸化位点时,往往返回近乎数百至数万个可能的翻译后修饰 (PTM) 位点或基序,数量级超过可能用于单独研究以确认激酶特异性者。事实上,已在人类蛋白质上鉴定出数万个丝氨酸/苏氨酸磷酸化位点,而数千者已知与人类疾病和生物过程有关 — 但鉴定到负责这些修饰的激酶不及 Ser/Thr 基序的 4%。
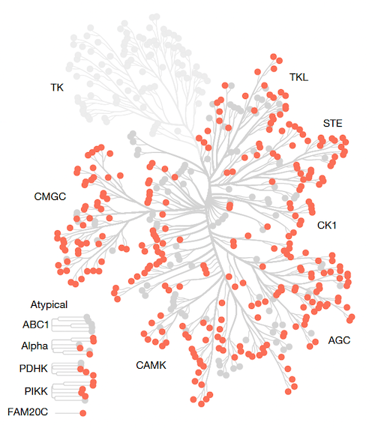
人类蛋白质激酶组进化树图,突出显示研究中分析的 Ser/Thr 激酶1。
然而,现代计算工具正被利用来提供传统技术无法获得的见解。通过访问正确的数据,Cantley 教授认识到可以开发这样一种预测算法,它利用蛋白质上磷酸化位点的结构预测哪种已知激酶会最可能与该蛋白质相互作用。但要创建这种算法,该团队首先需要确定大多数已知人类激酶的结合偏好性,以建立他们的预测模型。
纯化 Ser/Thr 激酶以确定结合偏好性
这项具有里程碑意义的事业始于 25 年前 Cantley 教授的实验室,他在此与这篇《自然》论文的其他资深作者——麻省理工学院精准癌症医学中心主任 Michael Yaffe 和耶鲁大学医学院药理学副教授 Benjamin Turk 共事。几十年来,三位资深人士一直在研究激酶及开创新方法阐明激酶的信号通路。
三人组首批突破之一是开发了一个肽文库,该文库为鉴定与各种激酶相互作用的基序而创建。这个基序文库由约 25 亿种肽底物组成,这些底物代表一个肽基序所有可能的氨基酸序列。为了更好地说明这一点,使用磷酸化作为示例,前提如下:通过以下方式构建肽文库:固定氨基酸序列内部的单个磷酸肽,然后变动这个固定的磷酸肽周围的氨基酸。结果就是这样一个“文库”,它包含可以与激酶相互作用的所有可能基序。接下来,使用目的激酶和放射性标记的 ATP (γ P32) 对肽文库进行体外磷酸化测定。这些肽阵列中的磷酸化程度可精确揭示激酶在其靶向的丝氨酸、苏氨酸或酪氨酸位点周围位置中偏好或排斥哪种氨基酸。
威尔康奈尔医学院药理学讲师 Jared Johnson 和威尔康奈尔医学院前研究生 Tomer Yaron 是这篇 《自然》论文的主要作者,负责确定纳入研究的激酶的底物结合偏好性。该团队利用 Cantley 教授构建的肽文库,优先确定了 303 种已知 Ser/Thr 激酶的底物结合偏好,这些激酶据预测占人类活性激酶的 84%。
这包括获取每种激酶的重组制备物并剖析它们在其肽阵列中的活性。通过这种方法,研究小组能够根据磷酸化位点周围的氨基酸序列,识别出激酶更倾向于磷酸化哪些底物。
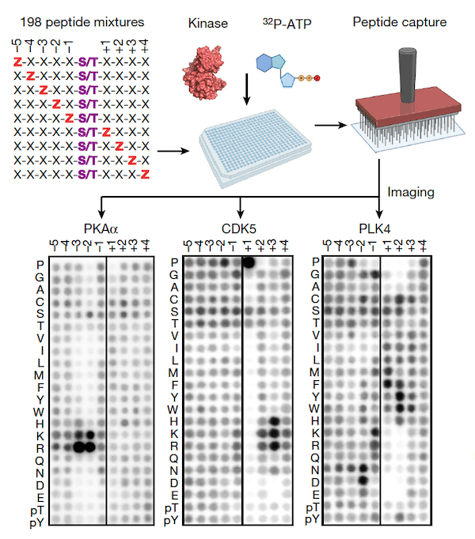 剖析人类丝氨酸/苏氨酸激酶组的底物特异性。PSPA 分析的实验工作流程和代表性结果。Z 表示包含 20 种天然氨基酸之一或磷酸化 Thr (pThr) 或磷酸化 Tyr (pTyr) 的固定位置。X 表示包含所有天然氨基酸(剔除 Ser、Thr 和 Cys)的随机混合物的非固定位置。较暗的斑点表示首选残基。使用 BioRender 创建示意图1。
剖析人类丝氨酸/苏氨酸激酶组的底物特异性。PSPA 分析的实验工作流程和代表性结果。Z 表示包含 20 种天然氨基酸之一或磷酸化 Thr (pThr) 或磷酸化 Tyr (pTyr) 的固定位置。X 表示包含所有天然氨基酸(剔除 Ser、Thr 和 Cys)的随机混合物的非固定位置。较暗的斑点表示首选残基。使用 BioRender 创建示意图1。
凭借这种关于每个已知 Ser/Thr 激酶的数据,开发了一个可以预测激酶底物结合偏好性的计算模型。为了评估每个激酶-肽对,研究人员利用按概率归一化并缩放的大型数据集,开发出一种可用于任何潜在底物氨基酸序列的评分指标。他们通过将激酶和给定底物肽之间的分数除以包含数万个位点的背景磷酸蛋白质组,把每个激酶-肽对的偏好性分数作为百分位数算出。偏好分数表示与细胞中所有其他潜在底物相比,给定激酶对特定底物的偏好程度。
“磷酸化蛋白质组学的复兴”
令研究人员惊讶的是,他们发现许多情况下具有完全不同氨基酸序列的蛋白激酶使得具有相似底物基序的蛋白质磷酸化。此外,该团队发现,大约一半已研究的 Ser/Thr 激酶靶向三大类基序之一。剩下的一半对大约不到十二个类别的基序有特异性。
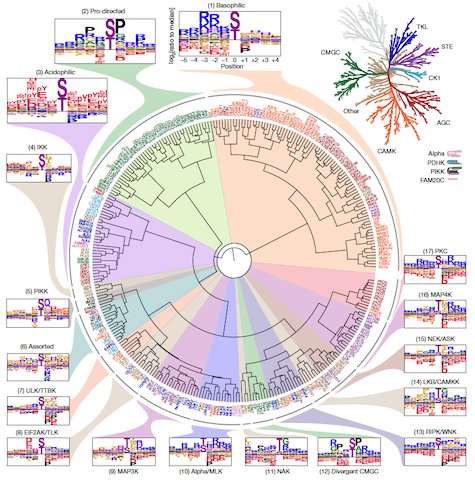
人类 Ser/Thr 激酶组的磷酸化位点基序树。303 种 Ser/Thr 激酶基于其氨基酸基序选择性 (PSSM) 的分层聚类。激酶名称根据其系统发育关系(右上图)作彩色标记2。阅读科学论文以查看全尺寸图像1。
为了检验他们的模型,该团队将其预测系统应用于已发布的非靶向磷酸化蛋白质组学数据集。在一个示例中,他们分析了来自细胞的数据,这些细胞经一种调节细胞生长的抗癌激酶抑制剂(名为 Plk1 )处理。仅根据已鉴定磷酸化位点的氨基酸序列且在不了解基础生物学情况下,该团队可以将 Plk1 确定为下调的磷酸化事件中最频繁预测到的激酶。此外,他们能够发现涉及额外激酶群组的更遥远下游通路,从而突显了他们的预测系统可以怎样将复杂的总体磷酸化蛋白质组学数据组织成信号转导通路。
该团队开发的计算模型现在托管在激酶文库的 Phosphosite 上,在那里,研究人员可以上传自己正在研究的一个或多个基序位点的序列并收到最有可能与该位点相互作用的激酶列表。
“凭借这个工具,您可以查看磷酸化的蛋白质并开始关联哪种激酶可能导致该磷酸化事件,”CST 研究执行总监 Sean Beausoleil 解释道。
该工具可用于探查导致疾病(包括癌症形成和其他疾病)的功能失调性信号转导通路。在上传关于因某种特定处理而变化的磷酸化事件的数据后,该工具可以自动确定导致磷酸化增加或减少的激酶。
“这真正是磷酸化蛋白质组学的复兴,”Sean 继续说道:“每个做过磷酸化实验的人都应该通过这个工具运行他们的数据集。”
其他资源
了解 PTMScan,CST 用于鉴定 PTM 的蛋白质组学抗体和试剂盒产品线,这些抗体和试剂盒和上述 Cantley 教授的25 亿种肽的文库工作同步开发。
参考文献
- Johnson JL, Yaron TM, Huntsman EM, et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature. 2023;613(7945):759-766. doi:10.1038/s41586-022-05575-3. CC BY 4.0.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912-1934. doi:10.1126/science.1075762
- Massachusetts Institute of Technology. Enzyme 'atlas' helps researchers decipher cellular pathways: Biologists have mapped out more than 300 protein kinases and their targets, which they hope could yield new leads for cancer drugs. ScienceDaily. ScienceDaily, 12 January 2023.
- Nita-Lazar A, Saito-Benz H, White FM. Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics. 2008;8(21):4433-4443. doi:10.1002/pmic.200800231
- Han X, Aslanian A, Yates JR 3rd. Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12(5):483-490. doi:10.1016/j.cbpa.2008.07.024


 沪公网安备31011502018823号
沪公网安备31011502018823号